Chapter 11 Assign Gene Signature
library(Seurat)
library(tidyverse)
library(magrittr)
library(data.table)11.1 Description
Given a gene list, calculate gene signature by averaging gene expresion
11.2 Load seurat object
combined <- get(load('data/Demo_CombinedSeurat_SCT_Preprocess.RData'))
Idents(combined) <- "cluster"11.3 Load gene lists, here using the layer-enriched genes as examples
f.ls <- list.files(
"data/GeneList/",
pattern = "FDR001.upDEG.csv$",
full.names = T,
recursive = T
)
names(f.ls) <-
f.ls %>% map(basename) %>% map( ~ str_remove(.x, "vs.*"))
layer.ls <- f.ls %>% map( ~ {
df <- fread(.x)
gn <- df$id[!is.na(df$id)]
gn <- gn[which(gn %in% rownames(combined))]
return(gn)
})11.4 Calcuate gene signature per gene list
mean.ls <- layer.ls %>% map_dfc(~ colMeans(x = as.matrix(combined@assays$SCT[.x, ]), na.rm = TRUE))
rownames(mean.ls) <- rownames(combined@meta.data)
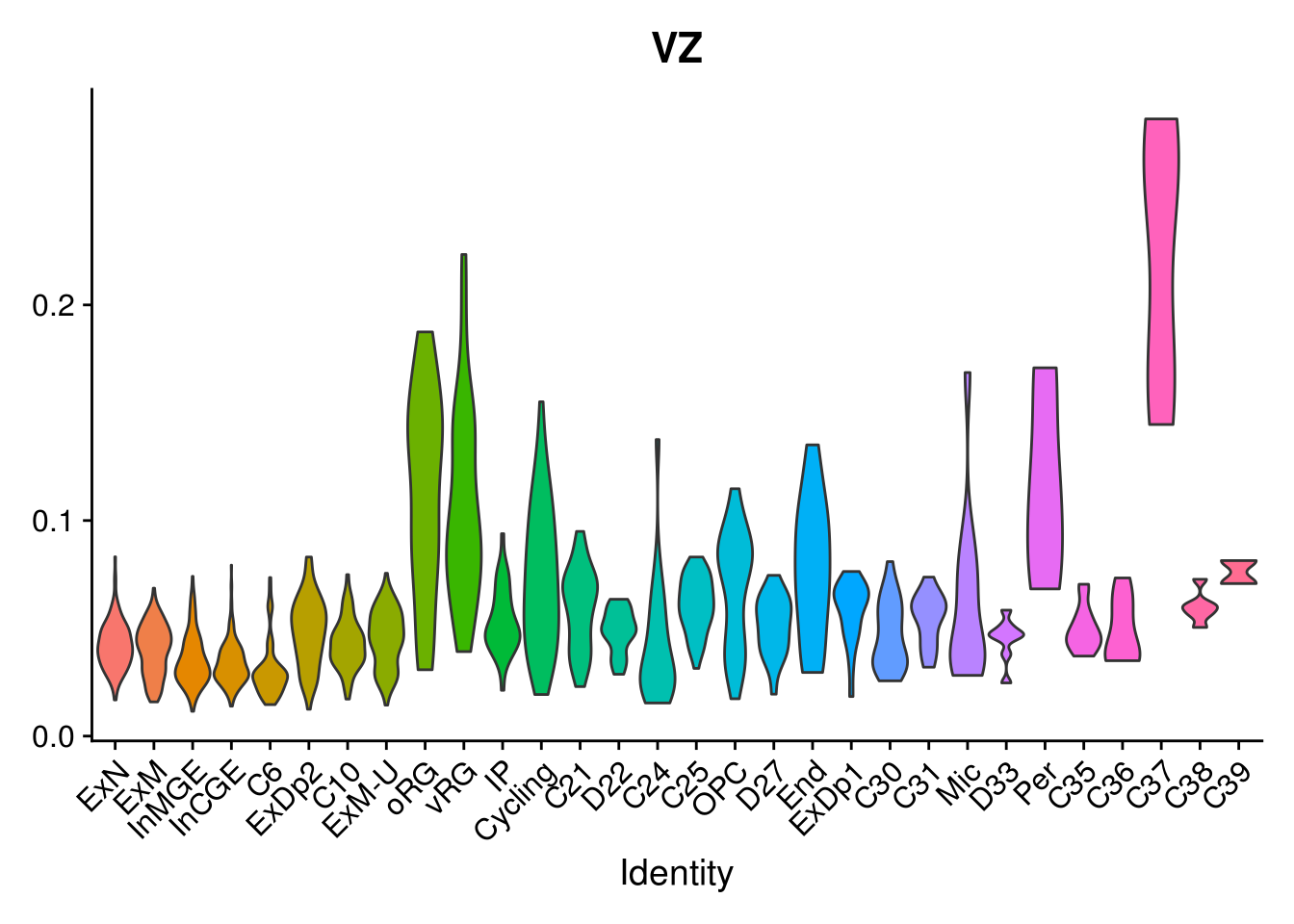
combined <- AddMetaData(combined, mean.ls, col.name = colnames(mean.ls))11.5 Explore the gene signature by FeaturePlot and VlnPlot
names(layer.ls) %>% map(~ FeaturePlot(object = combined, features = .x, pt.size = 0.001))## [[1]]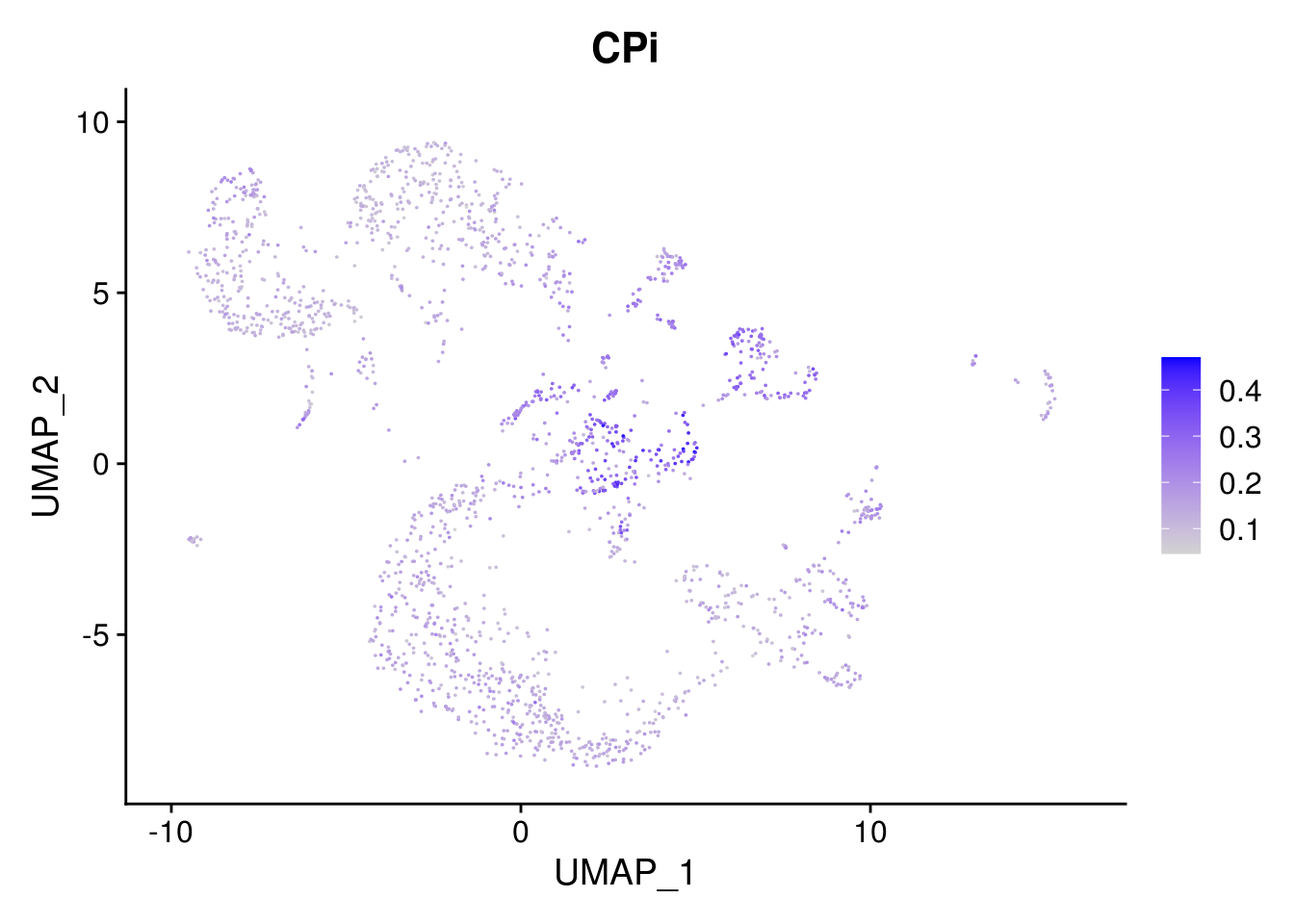
##
## [[2]]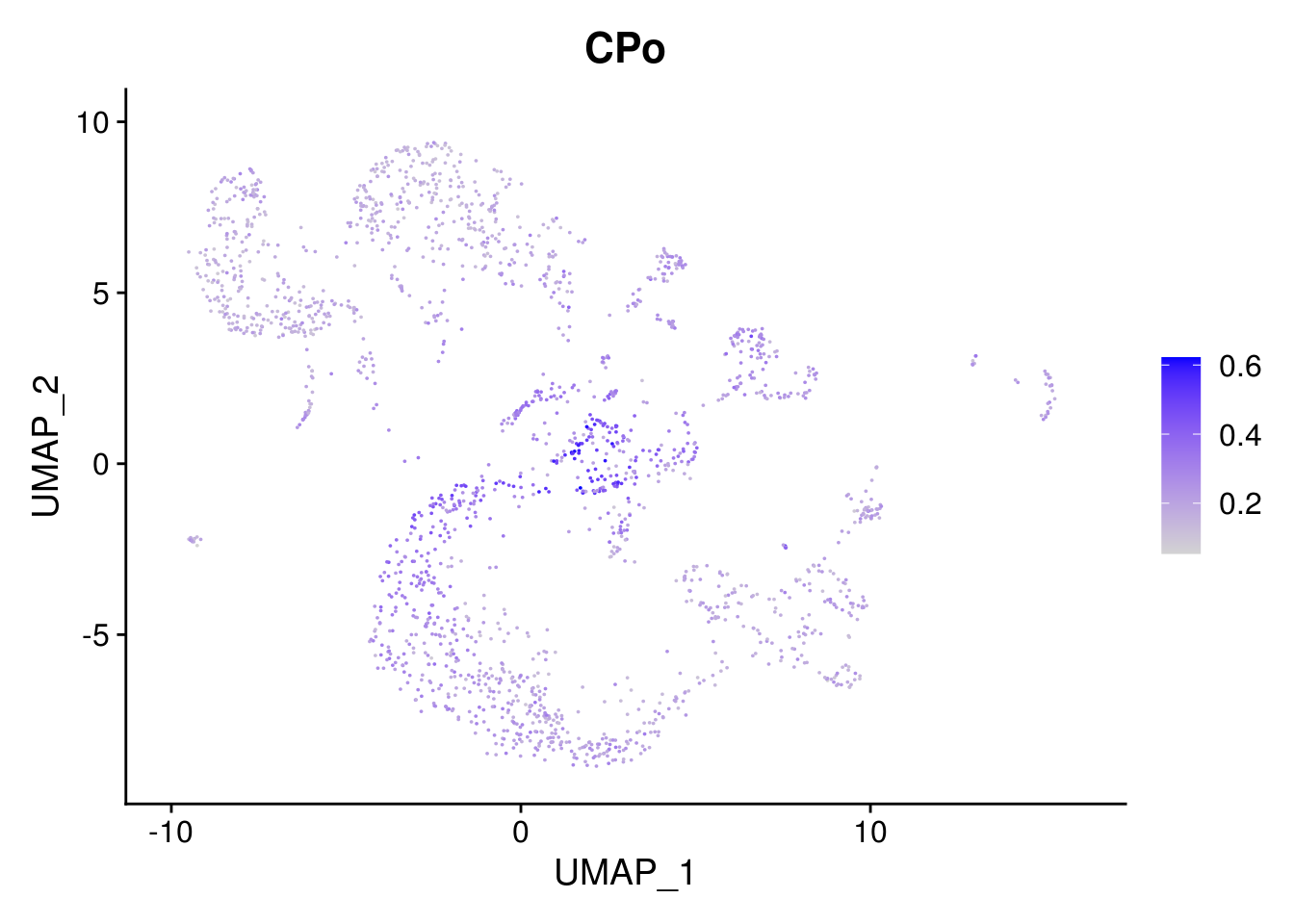
##
## [[3]]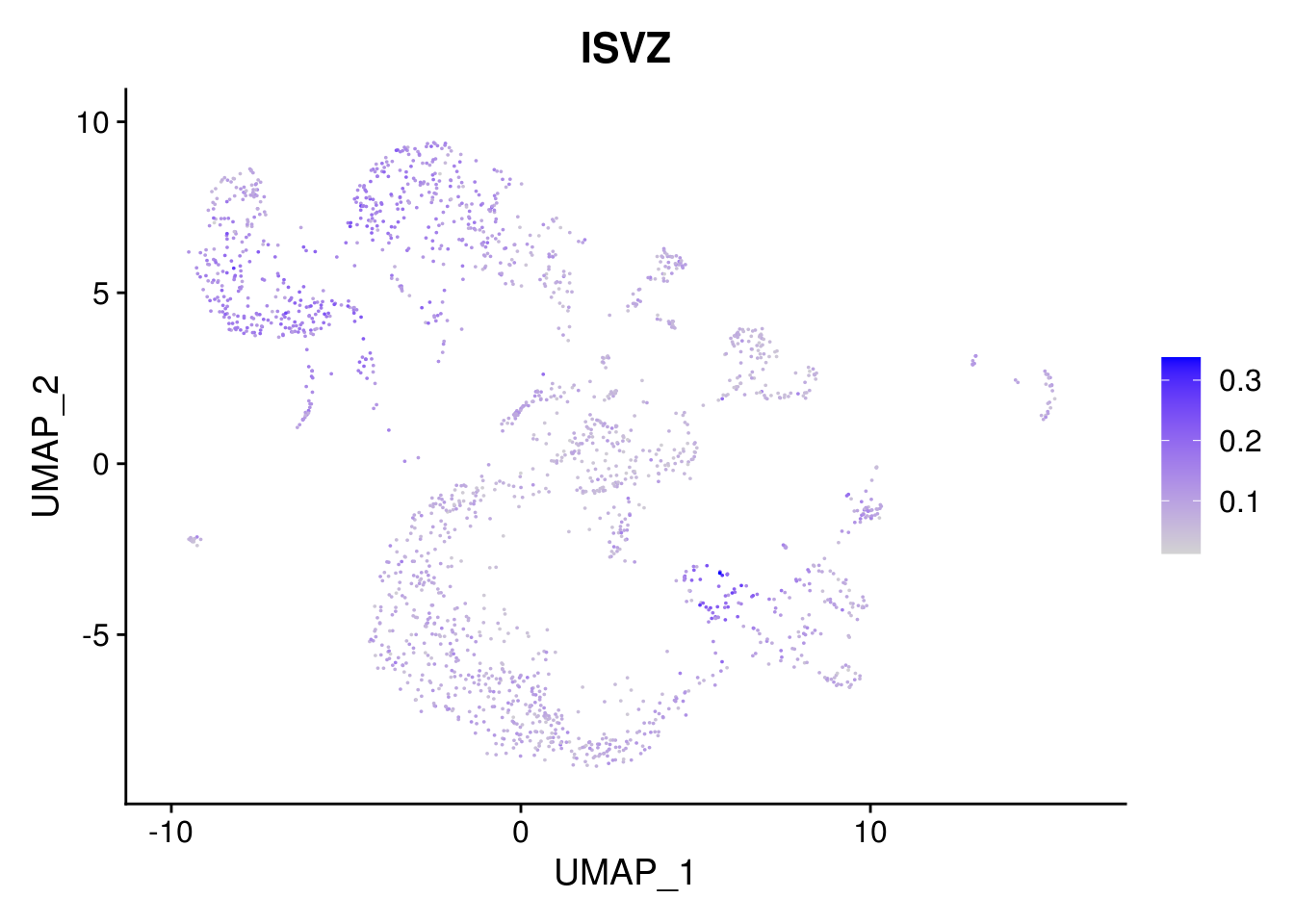
##
## [[4]]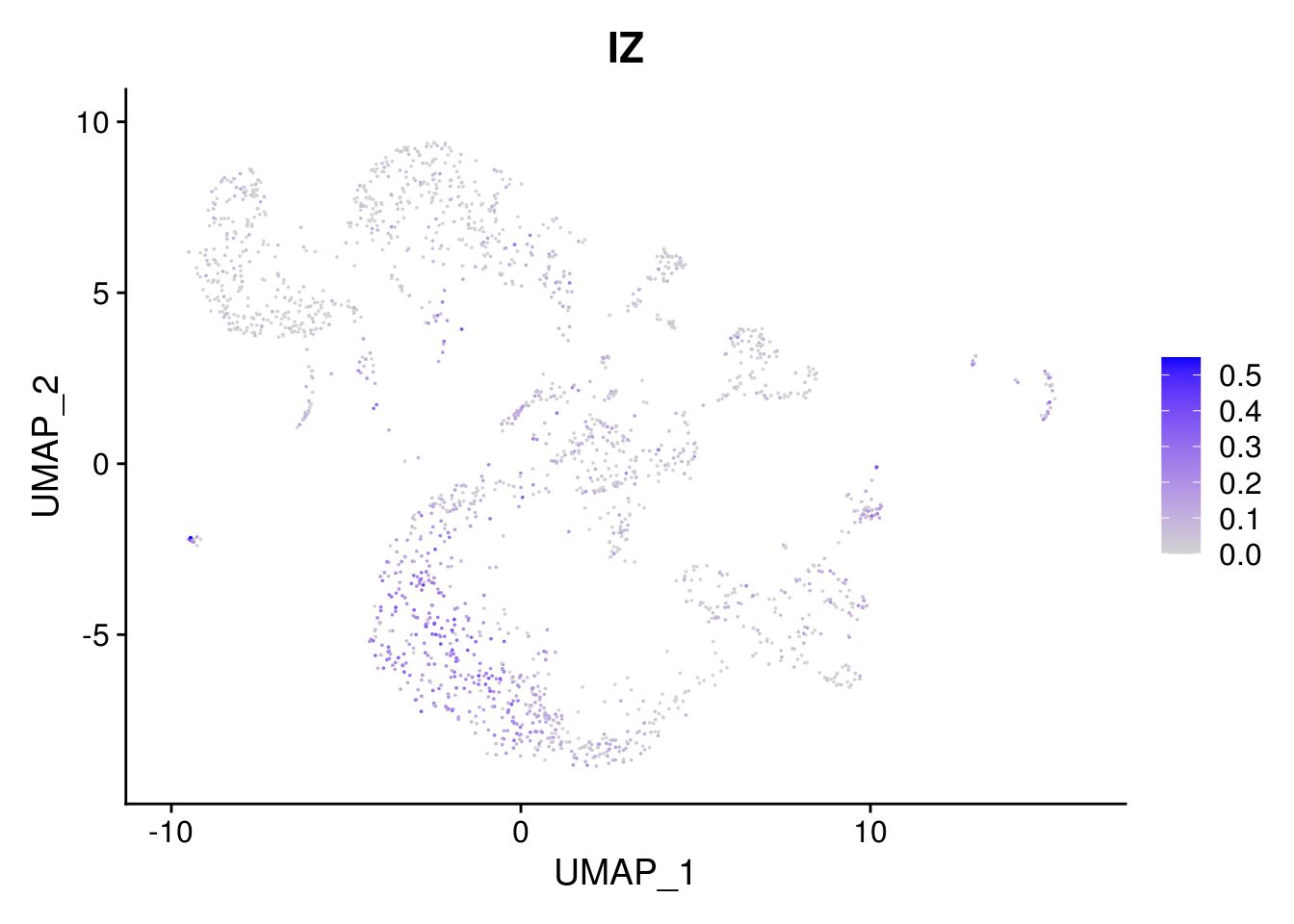
##
## [[5]]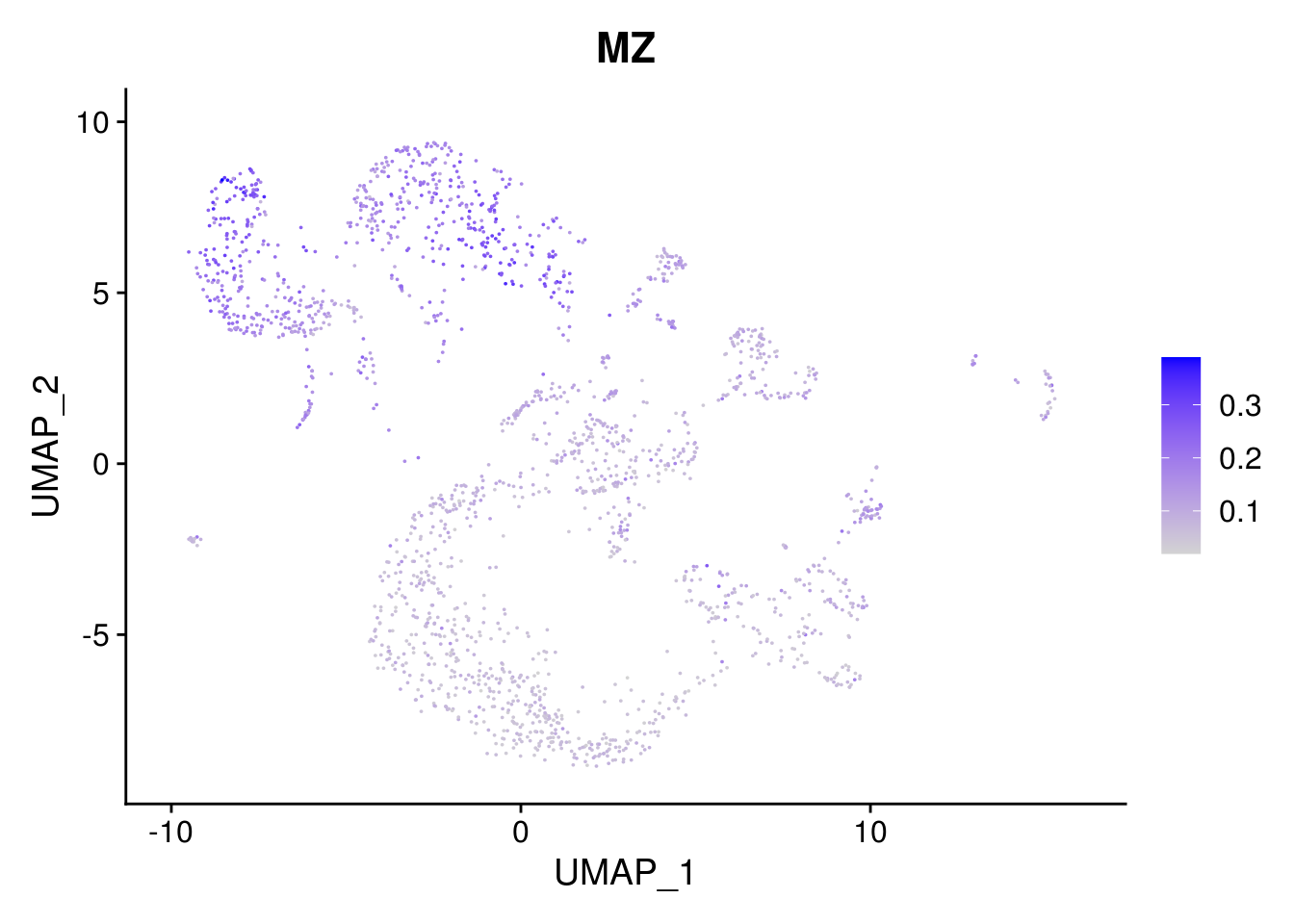
##
## [[6]]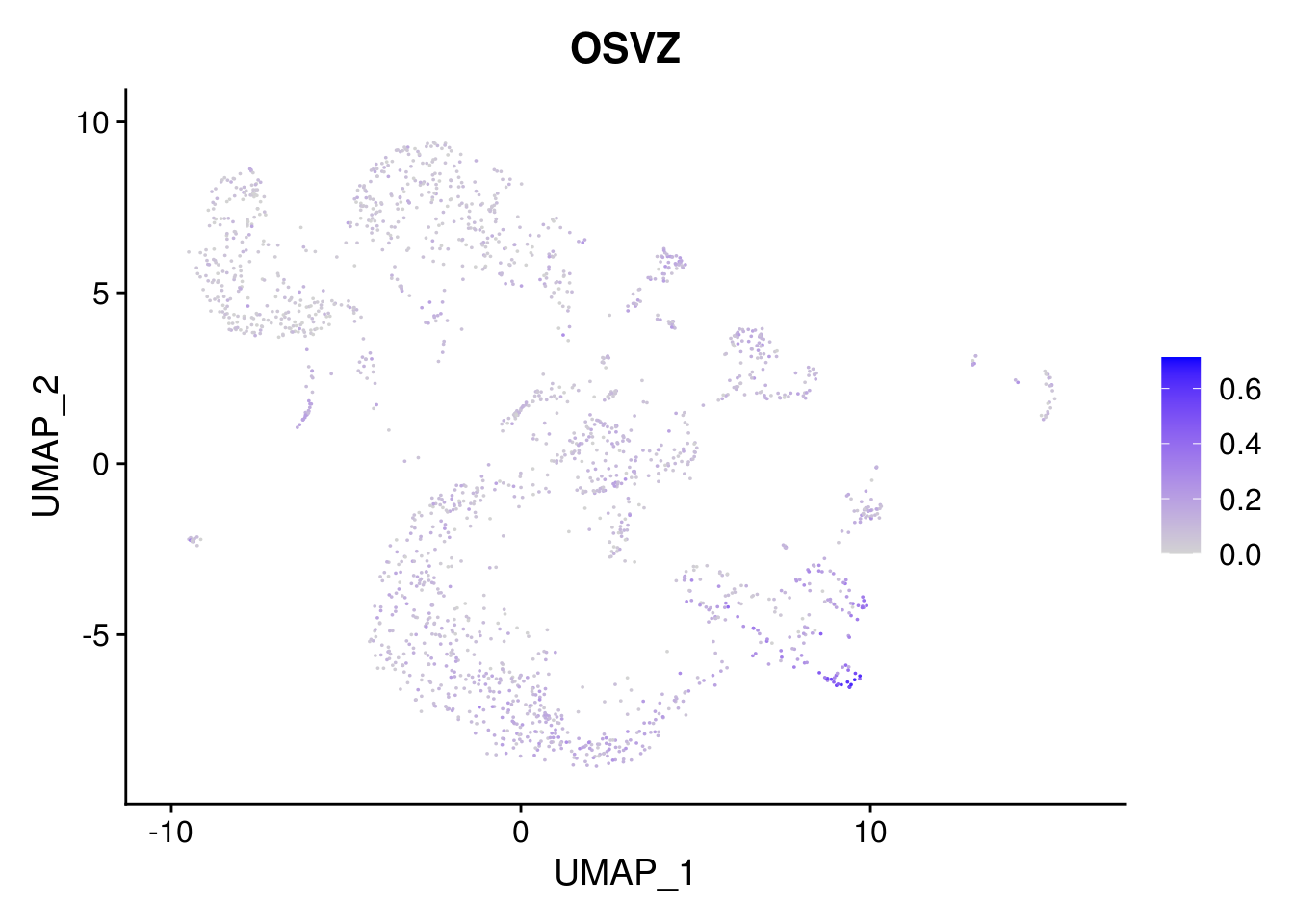
##
## [[7]]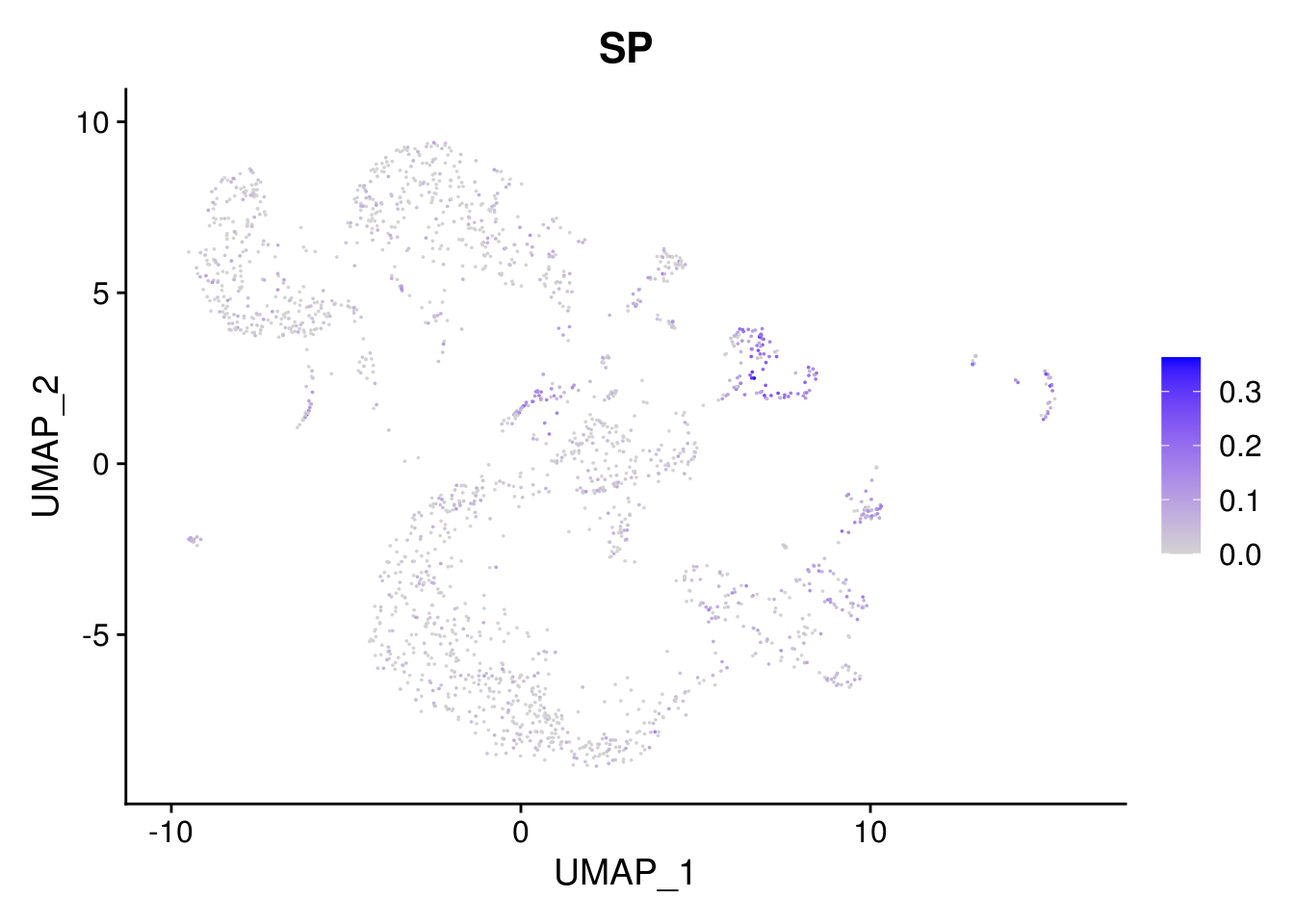
##
## [[8]]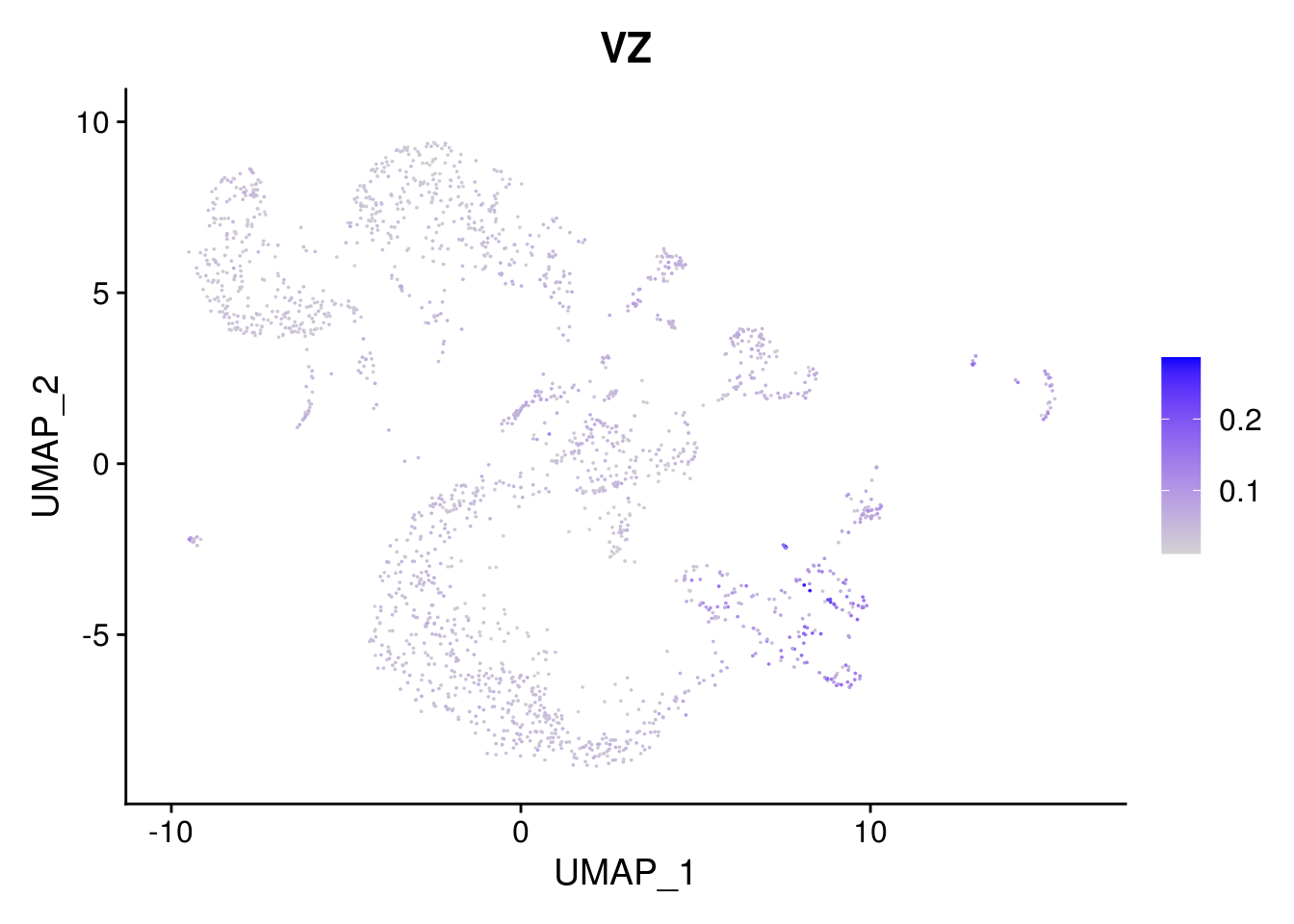
names(layer.ls) %>% map(~ {
VlnPlot(object = combined, features = .x, pt.size = 0) + NoLegend()
})## [[1]]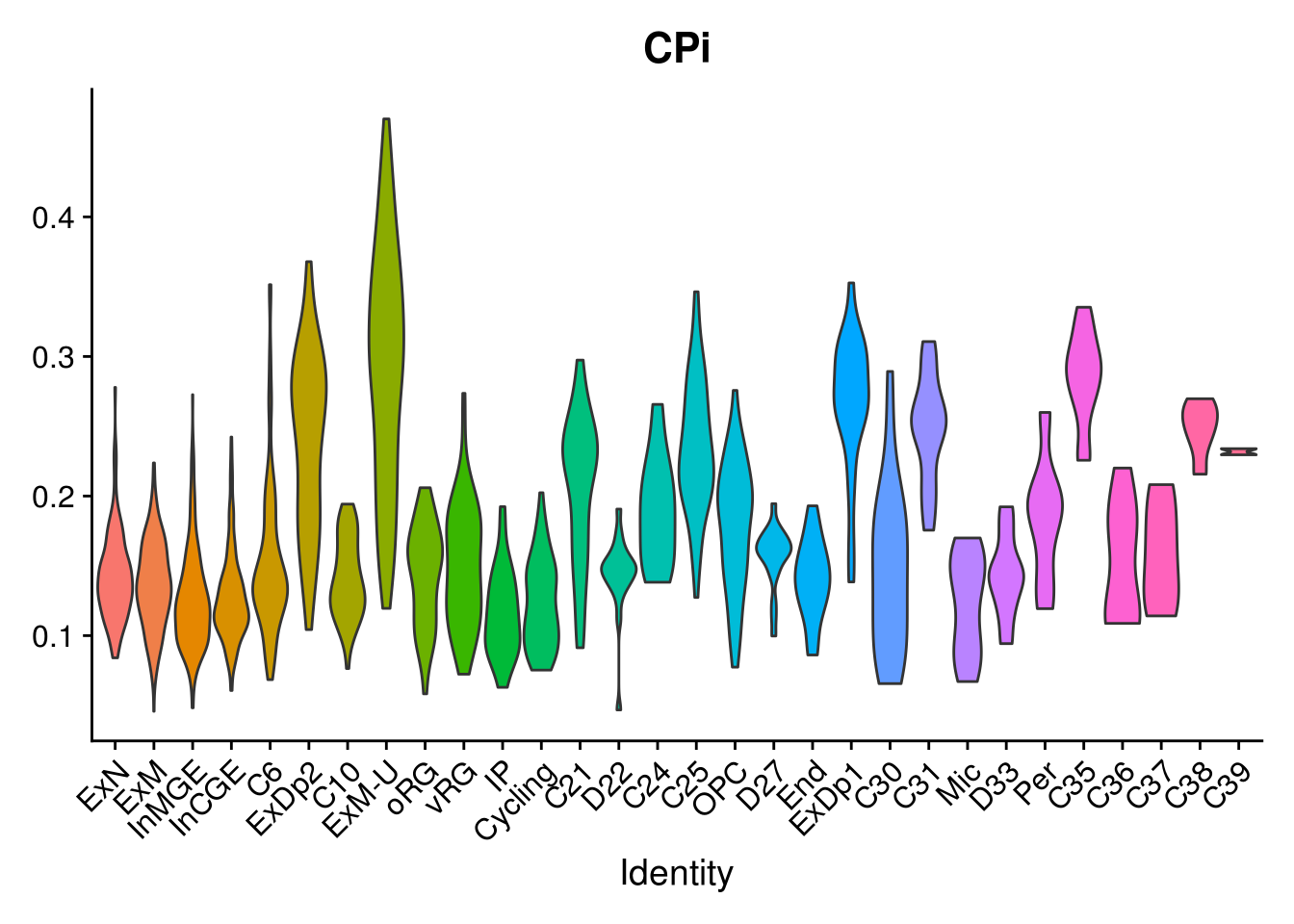
##
## [[2]]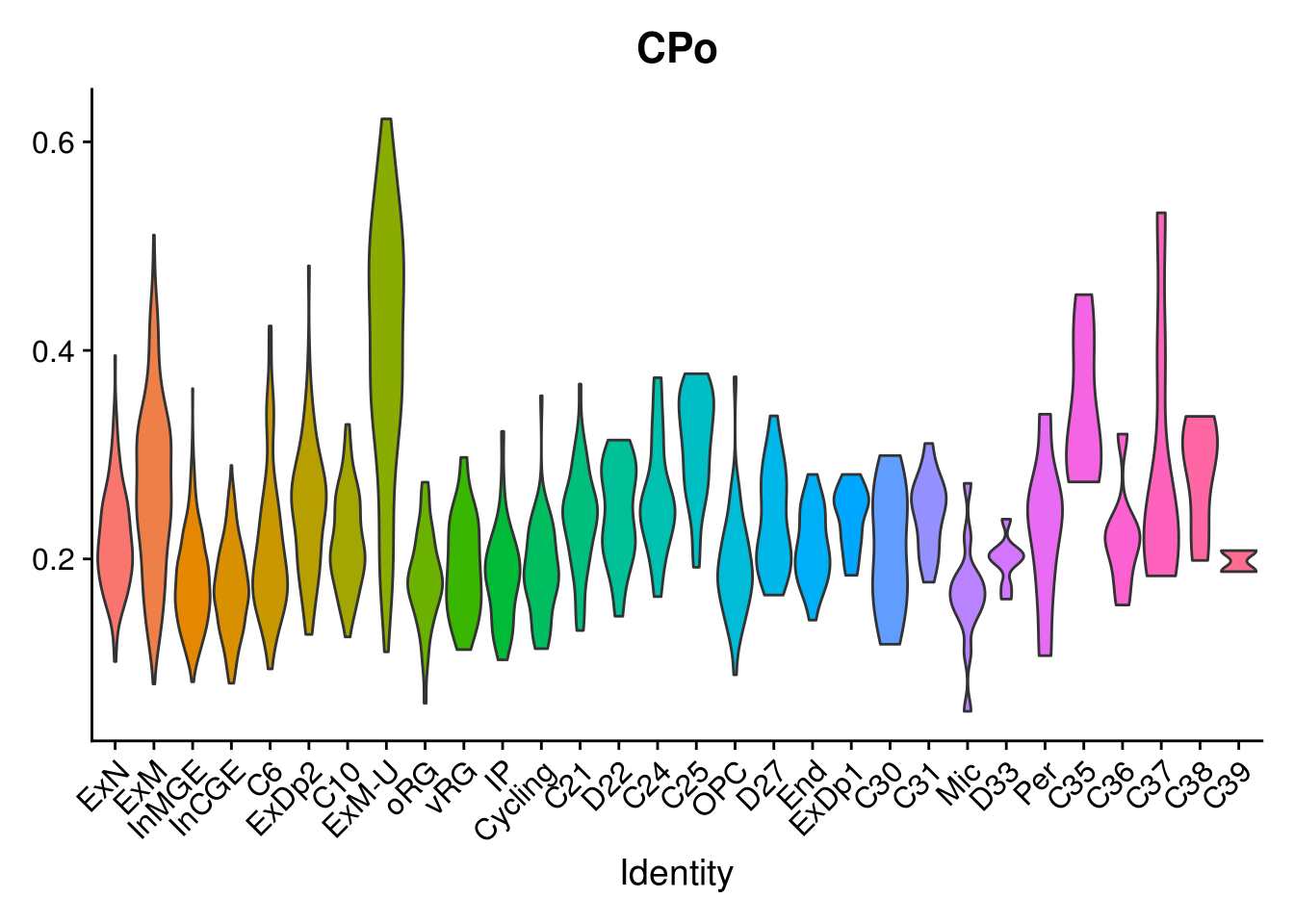
##
## [[3]]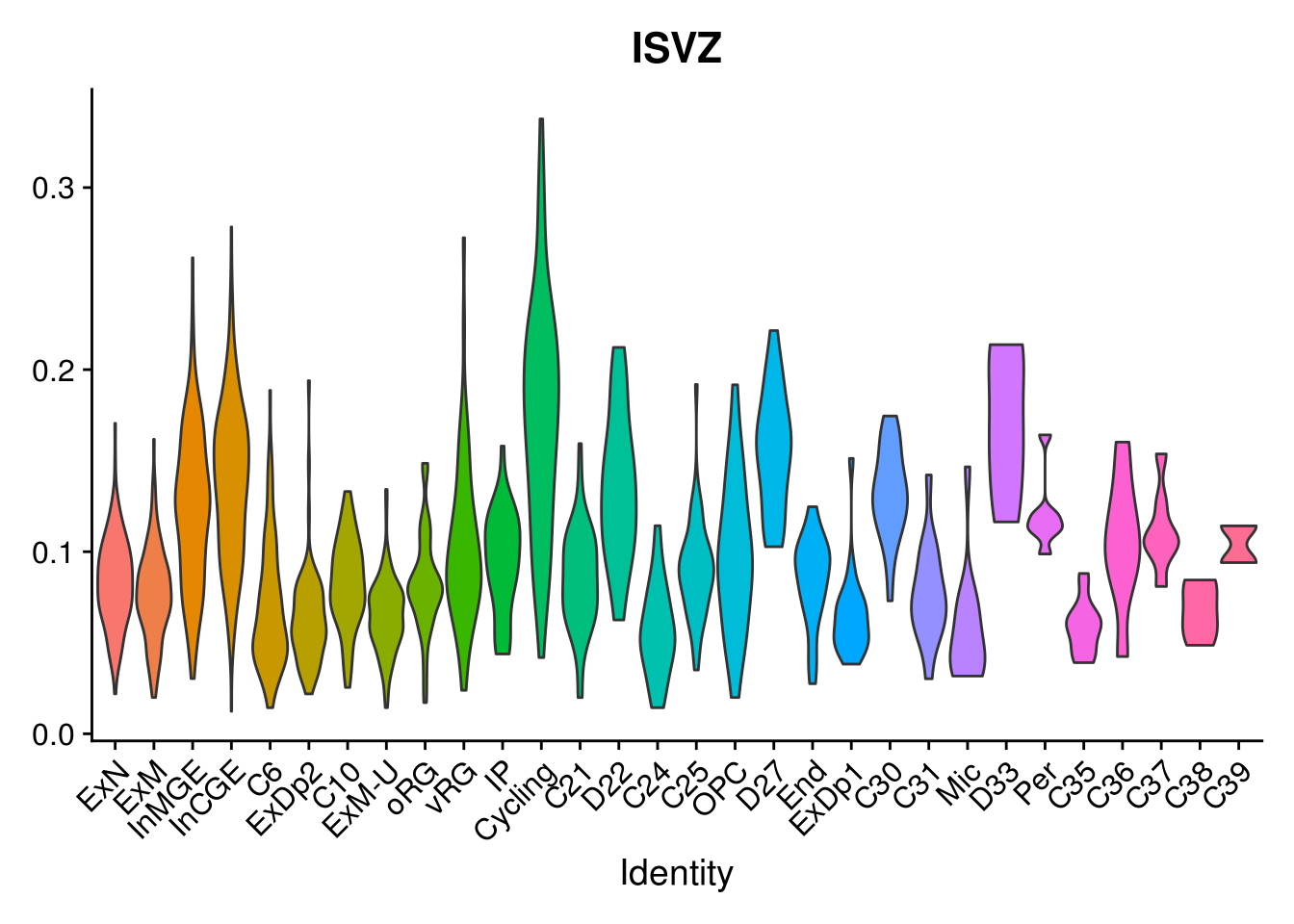
##
## [[4]]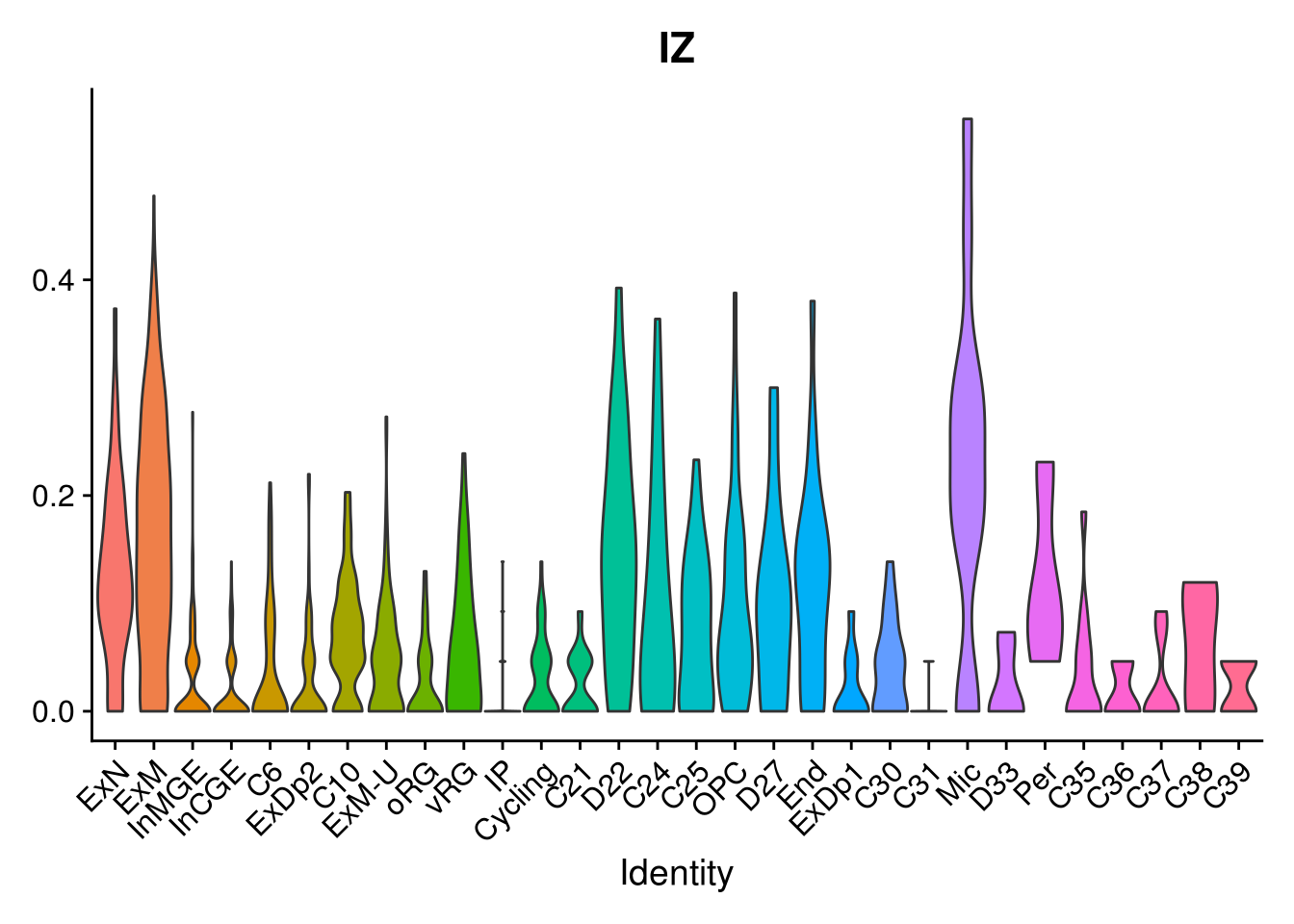
##
## [[5]]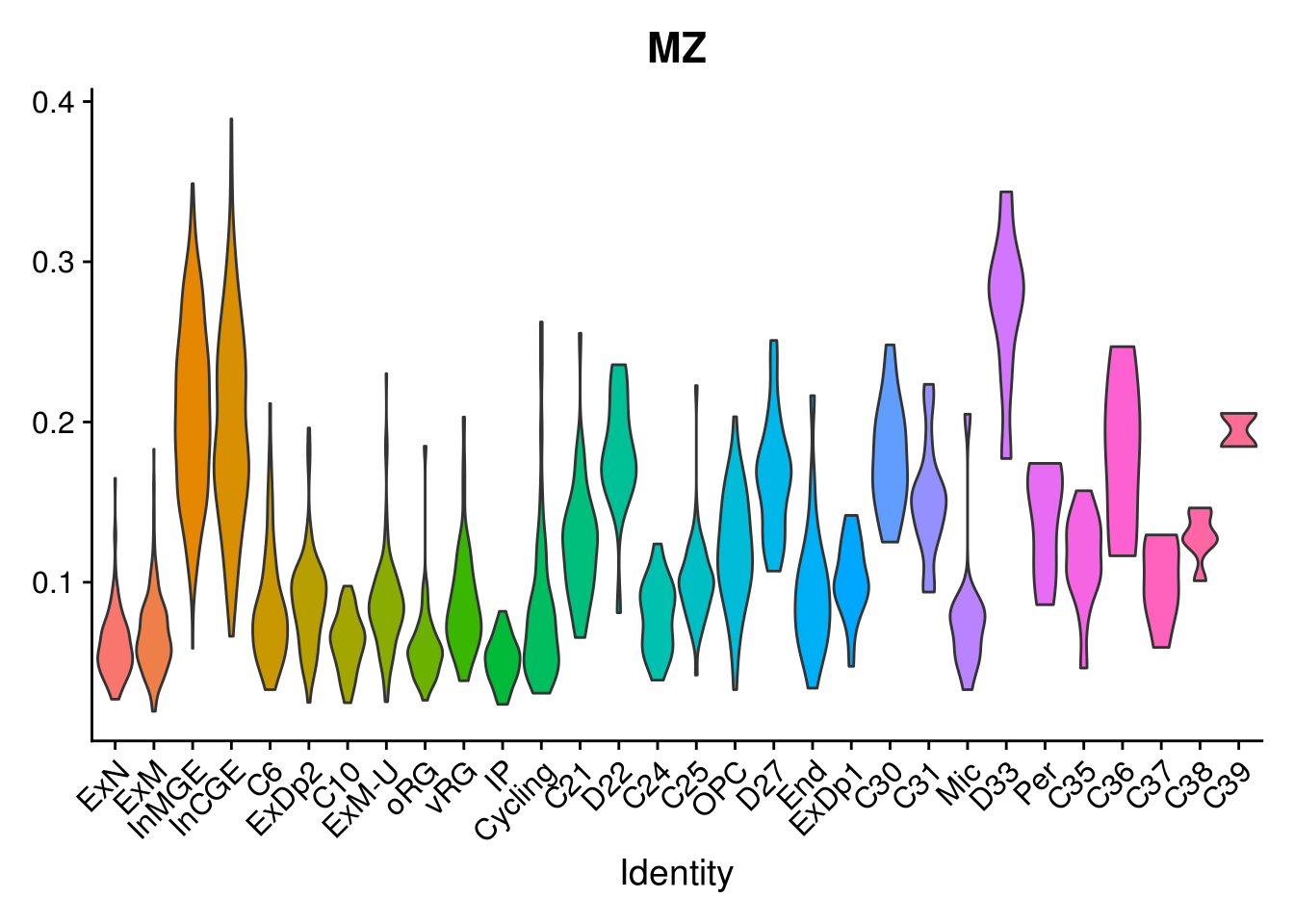
##
## [[6]]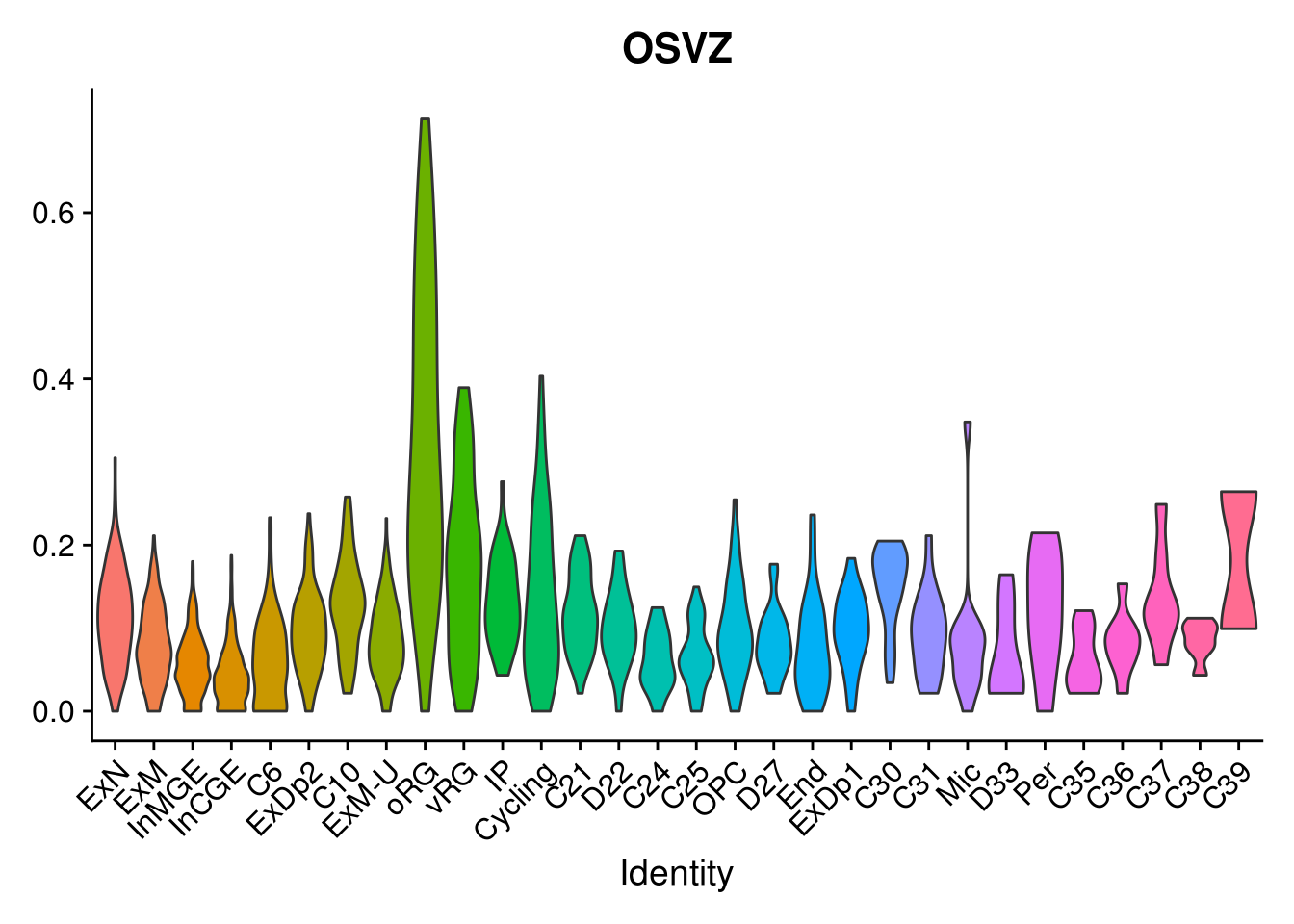
##
## [[7]]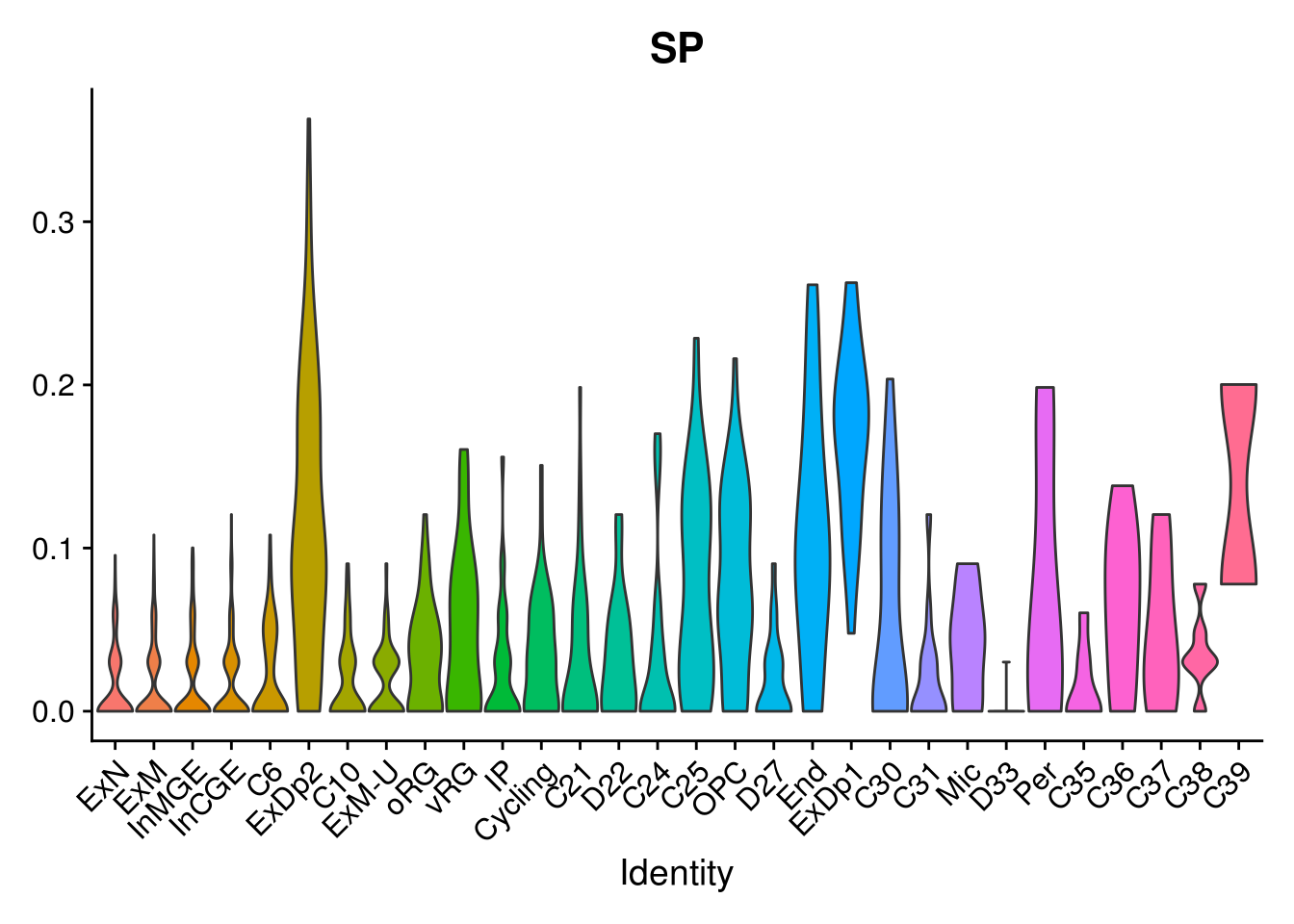
##
## [[8]]