7 Vraag7: airway
In les 6 is bij SummarizedExperiment the “airway” gen expressie dataset geïntroduceerd. Zoek informatie op over deze dataset en verzin 2 onderzoeksvragen met als uitgangspunt differentiële genexpressie. Vervolgens ga je met behulp van R de onderzoeksvragen beantwoorden. Geef duidelijk aan met comments waarvoor de R code dient en presenteer je resultaten met minimaal 1 relevante grafiek en 1 heatmap per onderzoeksvraag. Geef een beschrijving van je resultaten en een conclusie / discussie.
#BiocManager::install("airway")
#this causes problems when rendering the bookdown, no error message is displayed
require(airway)## Loading required package: airwaydata("airway")7.1 onderzoeksvraag 1:
Welke 10 genen hebben het grootste verschil in genexpressie tussen cellen de behandeld zijn met dexamethasone en zonder dexamethasone?
#calculate the average of the untreated groups and treated groups
treated <- as_tibble(assay(airway[,airway$dex == "trt"]))
untreated <- as_tibble(assay(airway[,airway$dex == "untrt"]))
treated<- treated %>% transmute(gene = BiocGenerics::rownames(assay(airway)),
mean_count_treated = rowMeans(treated))
untreated<- untreated %>% transmute(gene = BiocGenerics::rownames(assay(airway)),
mean_count_untreated = rowMeans(untreated))
mean_rna_count <- left_join(untreated, treated, by = "gene")
head(mean_rna_count)## # A tibble: 6 x 3
## gene mean_count_untreated mean_count_treated
## <chr> <dbl> <dbl>
## 1 ENSG00000000003 865 619.
## 2 ENSG00000000005 0 0
## 3 ENSG00000000419 523 547.
## 4 ENSG00000000457 250. 234.
## 5 ENSG00000000460 63.5 53.2
## 6 ENSG00000000938 0.75 0#calculate the difference of untreated vs treated
mean_rna_count <- mean_rna_count %>% mutate(difference = abs(mean_count_treated - mean_count_untreated))
#display the 10 highest genes
top_diff <- mean_rna_count %>% arrange(desc(difference)) %>% head(n=10)
knitr::kable(top_diff)| gene | mean_count_untreated | mean_count_treated | difference |
|---|---|---|---|
| ENSG00000087086 | 245990.25 | 144287.25 | 101703.00 |
| ENSG00000137801 | 40238.75 | 140851.25 | 100612.50 |
| ENSG00000108821 | 154459.25 | 58372.00 | 96087.25 |
| ENSG00000164692 | 250667.25 | 167974.00 | 82693.25 |
| ENSG00000103888 | 85479.75 | 155277.50 | 69797.75 |
| ENSG00000156508 | 293360.25 | 227849.75 | 65510.50 |
| ENSG00000168542 | 113389.25 | 55981.75 | 57407.50 |
| ENSG00000011465 | 328622.50 | 272857.50 | 55765.00 |
| ENSG00000115461 | 197575.50 | 145412.25 | 52163.25 |
| ENSG00000075624 | 55822.75 | 98929.25 | 43106.50 |
De tien genen die het meest verschil hadden in genexpressie voor en na behandeling met dexamethasone (n=4).
#display counts of the 10 highest genes, treated vs untreated
top_diff_tidy <- top_diff %>% pivot_longer(cols = 2:3,names_to = "DEX", values_to = "mean_count", names_prefix = "mean_count_")
top_diff_tidy %>% ggplot(aes(x = reorder(gene, difference),
y = mean_count,
fill = DEX)) +
geom_bar(stat = "identity", position = "dodge") +
theme_minimal() +
coord_flip() +
labs(title = "Hoogste verschil in expressie",
x = "Gen",
y = "gemiddeld RNA transcipt",
fill = "dexamethasone")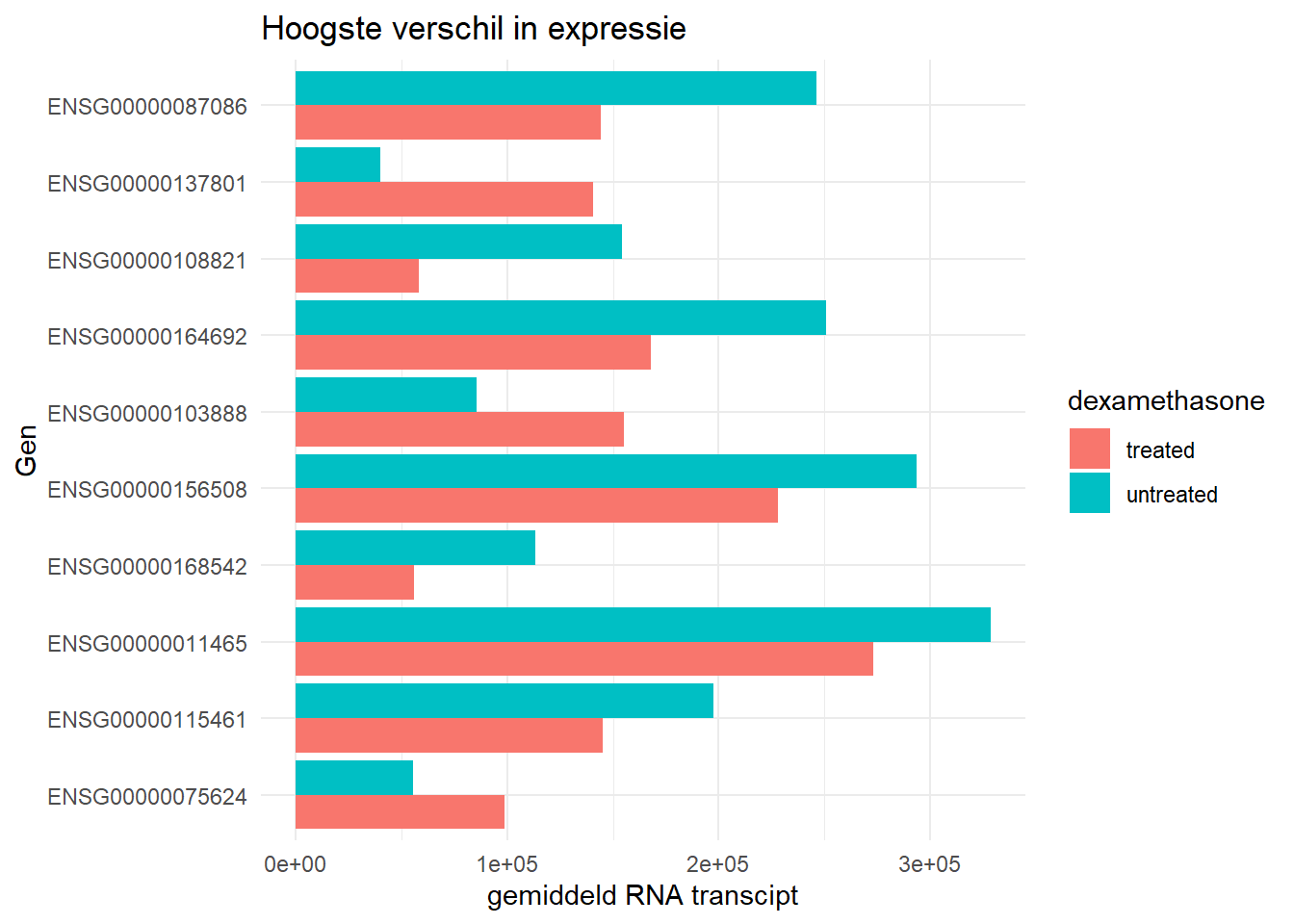
#plotting heatmap
top_diff_genes <- top_diff %>% dplyr::select("gene") %>% pull()
matrix_top_diff <- assay(airway[top_diff_genes,])
colnames(matrix_top_diff) <- colData(airway)[,"dex"] %>% str_replace_all(c("untrt", "trt"), c("untreated", "treated"))
pheatmap(matrix_top_diff,
main = "Heatmap van RNA transcript aantal
",
scale = "column")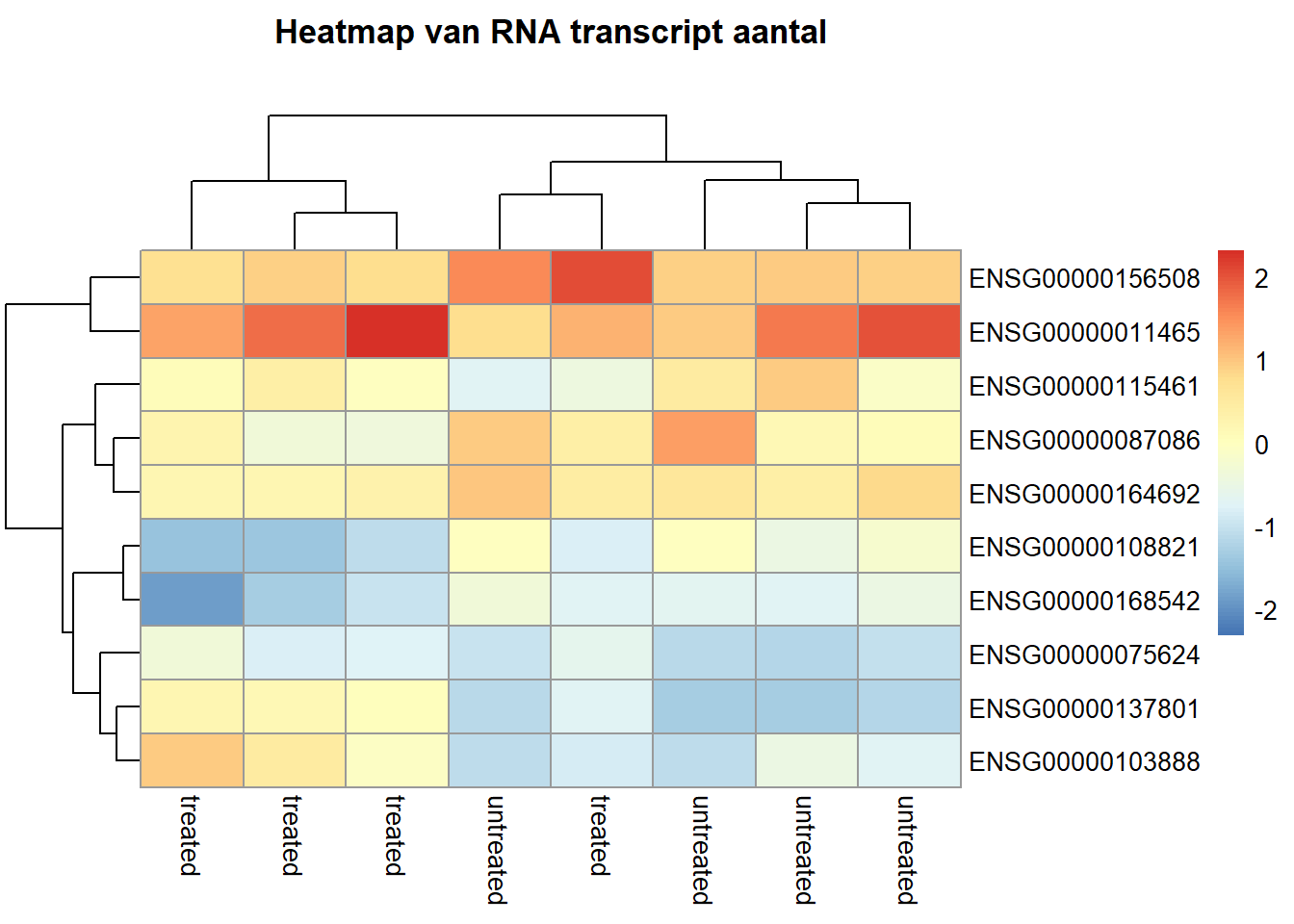 In de heatmap is terug te zien dat bepaalde genen over het algemeen een hogere expressie hebben dan andere.
In de heatmap is terug te zien dat bepaalde genen over het algemeen een hogere expressie hebben dan andere.
Conclusie De tien hoogste genen de het meeste van expressie zijn veranderd door de behandeling met dexamethasone zijn: ENSG00000087086, ENSG00000137801, ENSG00000108821, ENSG00000164692, ENSG00000103888, ENSG00000156508, ENSG00000168542, ENSG00000011465, ENSG00000115461, ENSG00000075624
Discussie Bij het analyseren van de data is er geen rekening gehouden met of het verschil in expressie positief of negatief is.
7.2 Onderzoeksvraag 2
Is er een verschil in expressie tussen de verschillende ASM-cellijnen zowel voor als na behandeling met dexamethasone?
# Maak een tibble met gemiddelde expressie niveaus per cellijn
mean_expression <- assay(airway) %>%
colMeans() %>% tibble(cell_line = c("N61311", "N61311", "N052611", "N052611", "N080611", "N080611", "N061011", "N061011"), treatment = c("untreated", "treated", "untreated", "treated", "untreated", "treated", "untreated", "treated"), mean_expression = .)
knitr::kable(mean_expression)| cell_line | treatment | mean_expression |
|---|---|---|
| N61311 | untreated | 321.9552 |
| N61311 | treated | 293.4305 |
| N052611 | untreated | 395.4424 |
| N052611 | treated | 236.5514 |
| N080611 | untreated | 381.3985 |
| N080611 | treated | 480.7684 |
| N061011 | untreated | 298.3706 |
| N061011 | treated | 330.1634 |
# Maak een grafiek van de expressie niveaus per cellijn
mean_expression %>% ggplot(aes(x = cell_line, y = mean_expression)) +
geom_col(aes(fill = treatment), position = "dodge") +
labs(title = "Gemiddelde expressie per cellijn",
subtitle = "Per behandeld en onbehandeld monster",
y = "Expressie", x = "", fill = "")
# Maak een heatmap van de data
expression_heat <- spread(mean_expression, key = cell_line, value = mean_expression)
expression_heat <- rbind(expression_heat$N61311, expression_heat$N052611, expression_heat$N080611, expression_heat$N061011)
colnames(expression_heat) <- c("treated", "untreated")
rownames(expression_heat) <- c("N61311", "N052611", "N080611", "N061011")
expression_heat## treated untreated
## N61311 293.4305 321.9552
## N052611 236.5514 395.4424
## N080611 480.7684 381.3985
## N061011 330.1634 298.3706pheatmap(expression_heat, main = "heatmap genexpressie per cellijn")